|
| |
Esta página contém maiores detalhes sobre o software Phrap.

 | Objetivos
O principal objetivo do Phrap é realizar a montagem de seqüências de
DNA em contigs (seqüências decorrentes da junção de duas ou mais
seqüências baseado em regiões de overlap entre elas). Se as
informações sobre a qualidade das seqüências lidas existir, estas são
usadas para melhorar o desempenho no processo de montagem.
Além da montagem, o Phrap também produz diversas informações sobre os
contigs gerados. Esses dados são úteis no processo de visualização do
resultado e no processo de finalização da montagem de fragmentos, onde
geralmente a análise humana irá tomar decisões que não puderam ser
realizadas automaticamente.
|
|
Interface
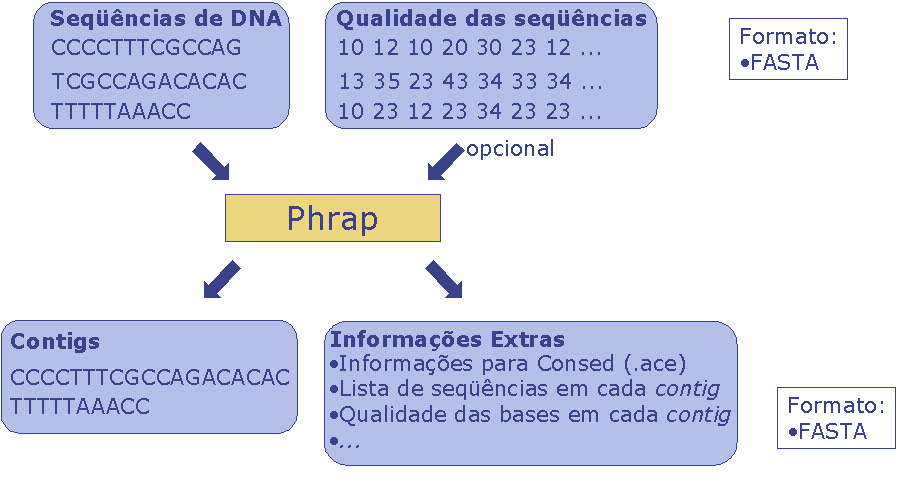
A seguir é ilustrada a comunicação com o software Phrap. Basicamente
ela se dá via arquivos, que deve ser no formato FASTA, tanto de entrada
quanto de saída. Observe que os
dados de entrada são aqueles produzidos pelo Phred, e que os de saída são
os recebidos pelo Consed. A figura seguinte
ilustra essa troca de dados.
 |
| Algoritmo
A seguir está uma descrição dos passos do algoritmo utilizado pelo
Phrap para definição de Contigs.
1. Realiza um pre-processamento da entrada;
2. Determina regiões de overlap entre todos os pares de seqüências;
3. Cria um grafo direcionado baseado no passo 2;
4. Produz contigs baseado no grafo definido em 3;
A definição dos contigs (passo 4) utiliza o algoritmo guloso para
selecionar as arestas do grafo em ordem decrescente .
O pre-processamento da entrada (passo 1) contém os seguintes passos:
1. Constrói os complementos das seqüências lidas (do arquivo “nome.fasta”)
e adiciona ao conjunto de seqüências;
2. Elimina do conjunto as seqüências duplicadas; (a similaridade entre
todos os pares já é calculada aqui)
3. Faz um vector screening (ver abaixo) no conjunto de seqüências;
4. Salva o resultado num arquivo FASTA; Esse novo arquivo (nomeado “nome.fasta.screen”)
será o arquivo utilizado pelo phrap; (um novo arquivo de qualidade,
nomeado “nome.fasta.screen.qual” é também criado)
O vector screening (passo 3, acima) no conjunto de seqüências consiste em encontrar no conjunto de seqüências as seqüências de bases que
correspondem a vectors. Tais bases são modificadas para ´X´ e não serão
utilizadas pelo Phrap. Essas bases foram introduzidas em laboratório para a
geração dos cromatogramas, portanto não fazem parte do organismo em
estudo.
Observação: Os vectors a serem procurados, que são seqüências normais
(acgt...), devem estar em um arquivo no formato FASTA e ser fornecido como
parâmetro para o Phrap. Caso este arquivo não seja informado, o phrap
utiliza um arquivo padrão que contém todos os possíveis vectors usados
normalmente.
|
| Exemplo
A abordagem utilizada para esse exemplo de execução do Phrap foi: (i)partir
de uma seqüência conhecida; (ii)dividir essa seqüência em partes;
(iii)executar o Phrap dando como entrada essas partes e observar a
qualidade do resultado. Nesse exemplo, foram exploraradas seqüências com
repetições e não foram utilizados arquivos de qualidade, uma vez que as
seqüências foram editadas manualmente.
A seqüência original, abaixo ilustrada por uma figura, contém algumas
partes repetidas (de cor verde) inseridas entre partes não repetidas:

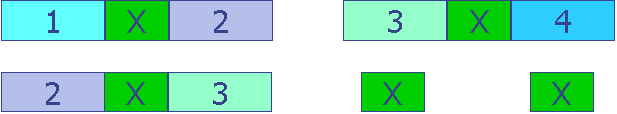
A partir dessa seqüência, 5 seqüências foram criadas e foram então
usadas como entrada para o Phrap. Essas 5 seqüências são ilustradas pela
figura abaixo.
O resultado obtido após a execução do Phrap foi excelente. Ele removeu as
seqüências duplicadas, e montou como esperado todas as seqüências. A
figura abaixo mostra o resultado.
|
|
Parâmetros de execução
Ao executar o Phrap, 53 parâmetros podem ser especificados de acordo com a
necessidade do usuário. Por exemplo, qualidade padrão para cada base (caso
não haja arquivo de qualidade), scores usados no alinhamento de
seqüências (mismatch, insertion, deletion, etc), tamanho mínimo de
overlap entre seqüências para que haja alinhamento, etc. |
|
